
Measuring how genome-wide diversity matters to threatened species has been a constant endeavor of conservation genetics, and still is in the era of genomics. The main idea still goes back to the small population paradigm that demonises low genetic diversity as it can lead to genetic drift and inbreeding, and hence lower population size in the following generations and imminent extinction. As I discuss in my previous post, this is not always the case.
Dissecting genome-wide diversity studies
There are four papers that came to my attention that includes an investigation on how genome-wide diversity relates to the threat status of species of conservation concern:
- Diez-del-Molino et al (2018) which found that the threat status of the species does not correlate well with their genome-wide diversity.
- Westbury et al (2018) who seek to figure out how low the brown hyenas are in genome-wide diversity, so that they include raw sequence data of several other taxa
- Bruniche-Olsen et al (2018) who measure the number of segregating sites and compare it between threatened and non-threatened taxa and find no significant difference
- Zoonomia Consortium (2020) found that overall heterozygosity declines with the level of threat of the species they sequenced.
What happened here?
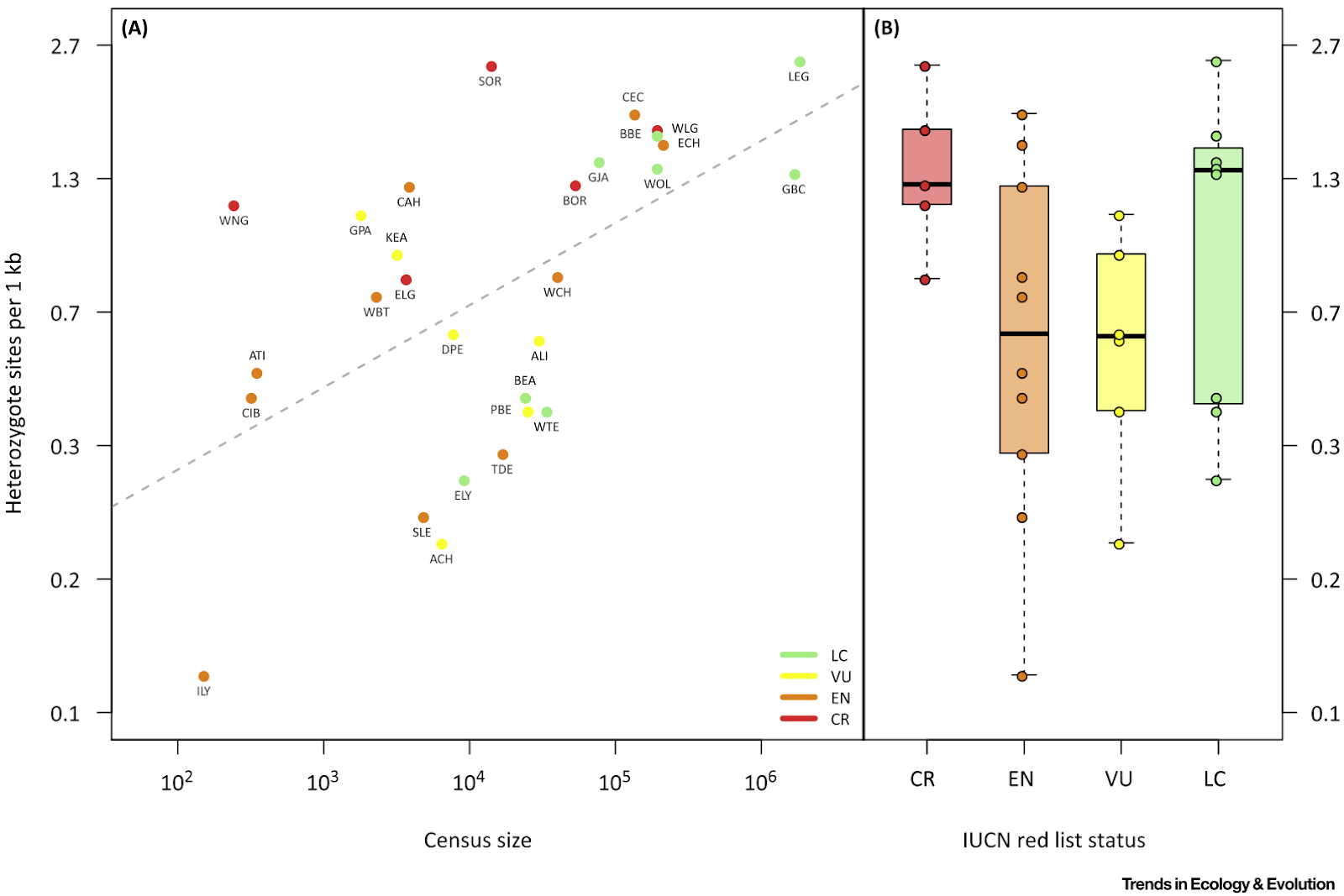


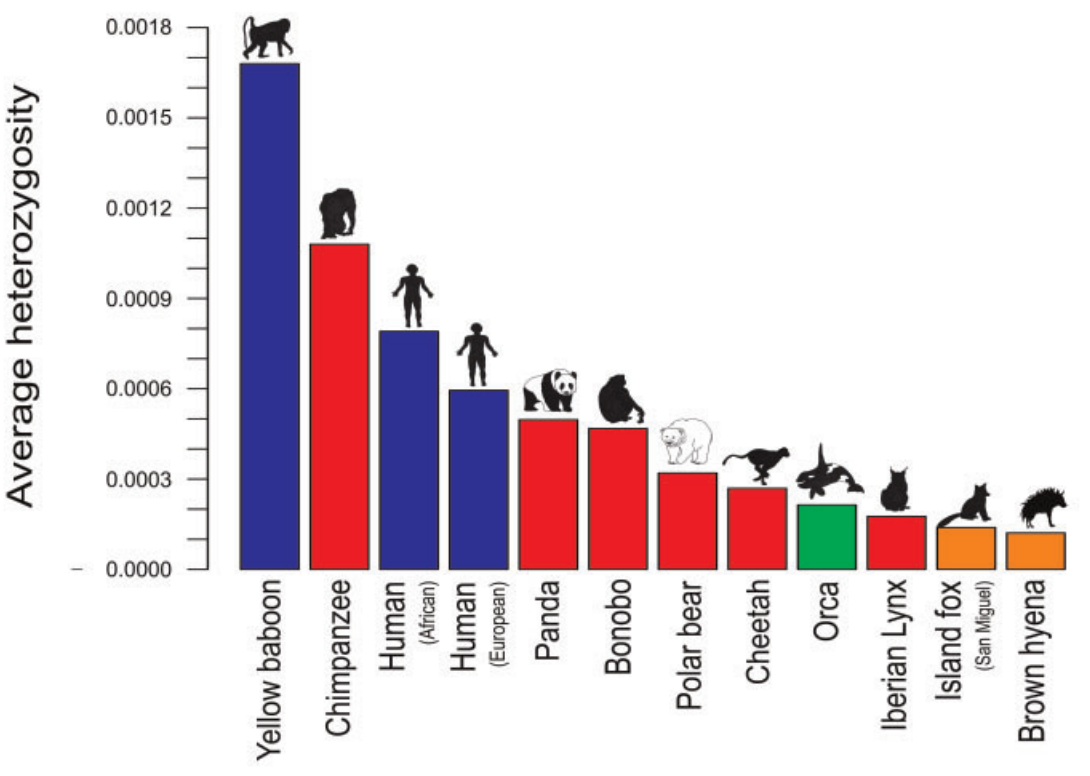
If you investigate each paper, you can see some notable differences (Table 1). The Diaz-del-Molino paper used published estimates, while both the Westbury and Zoonomia papers use the same pipeline for their whole dataset, with the Zoonomia paper discarding some samples along the way due to quality issues. In terms of statistical analysis, only the Westbury paper does not try to formally test any relationship between threat status and overall heterozygosity as the authors are only interested in the genome-wide diversity of the brown hyena compared to other threatened taxa. The other two papers are using different hypothesis testing frameworks but model-wise, they have the same linear model equation. I cannot really be sure if it is the lack of samples or the lack of categories that makes the relationship different between studies as they were using different numbers of IUCN Red List categories. The Bruniche-Olsen paper found that the lower genomic diversity in the threatened carnivores is significant, although the IUCN category per se does not look like having much effect. Nonetheless, the latter paper is again not comparable with other studies as it uses a different categorisation of the IUCN Red List status. It goes without saying that if you want to include genetic diversity in IUCN assessments, you need to care for a lot of things.
| Study | Diez-del-Molino et al (2018) | Westbury et al (2018) | Bruniche-Olsen et al (2018) | Zoonomia Consortium (2020) |
| #taxa | 30 | 12 | 78 | 101 |
| Threat status | 4, CR, EN, VU, LC | 4, EN, NT, LC, DD | 2, non-threatened (LC, NT), and threatened (VU, EN, CR) | 5, CR, EN, VU, NT, LC |
| How genomic diversity obtained | Published papers | Averaging ANGSD heterozygosity estimates from sliding windows | Manually counted from GATK result | Manually counted from gVCF file (made with GATK) using custom script |
| How genomic diversity visualised | Heterozygous sites per 1kb | Average genome-wide heterozygous sites | log theta | Fraction of heterozygous calls over the whole callable genome |
| Threat status x genomic diversity test | ANOVA (F = 2.06, P = 0.14) | NA | Linear regression using log-transformed theta and multiple response variable (P=0.46 for IUCN category) | Linear regression using the IUCN category as an ordinal predictor (P = 0.011, R2= 0.064) |
Technicalities aside, I think many will agree that what makes species endangered does not necessarily make their genetic diversity lower or higher. This debate is actually quite old; Avise mentioned the issue of heterozygosity-related fitness in Molecular Markers, Natural History, and Evolution, published in 1994. There are also other papers that found that there is no trend between threat status, which is usually measured by census population size, and genome-wide heterozygosity, such as the work of Prado-Martinez et al (2013) in various wild ape populations. Another work, Romiguier et al (2014), more explicitly showed how genetic diversity is more affected by life history traits than population size. I think the Bruniche-Olsen paper which has attempted to include the notion of trophic level by separating carnivores from non-carnivores in their assessment kinda come close to consider life history traits.
So what do we make of genome-wide diversity?
A not-so-simple story of genome-wide diversity
A perspective paper I found recently discussed on Twitter was actually covering this issue. Here is their say:
“Nonetheless, supporting empirical evidence for the existence of a causal relationship between genetic diversity and population viability or adaptive potential is weak. If genetic diversity is indeed a major factor affecting the health and survival of populations in the wild, then one would expect endangered species to show, on average, lower levels of diversity. However, the International Union for Conservation of Nature (IUCN) Red List status is only a poor predictor of a species’ genome-wide nucleotide diversity (18–21) (Fig. 1) It has been previously argued that this lack of correlation reflects a deficient classification and that genome-wide patterns of neutral diversity should be incorporated into IUCN’s listing criteria to more accurately assess the likelihood of future extinction and the associated need for conservation (20, 22, 23).”
Teixeira and Huber (2021)
Here is their Fig. 1:
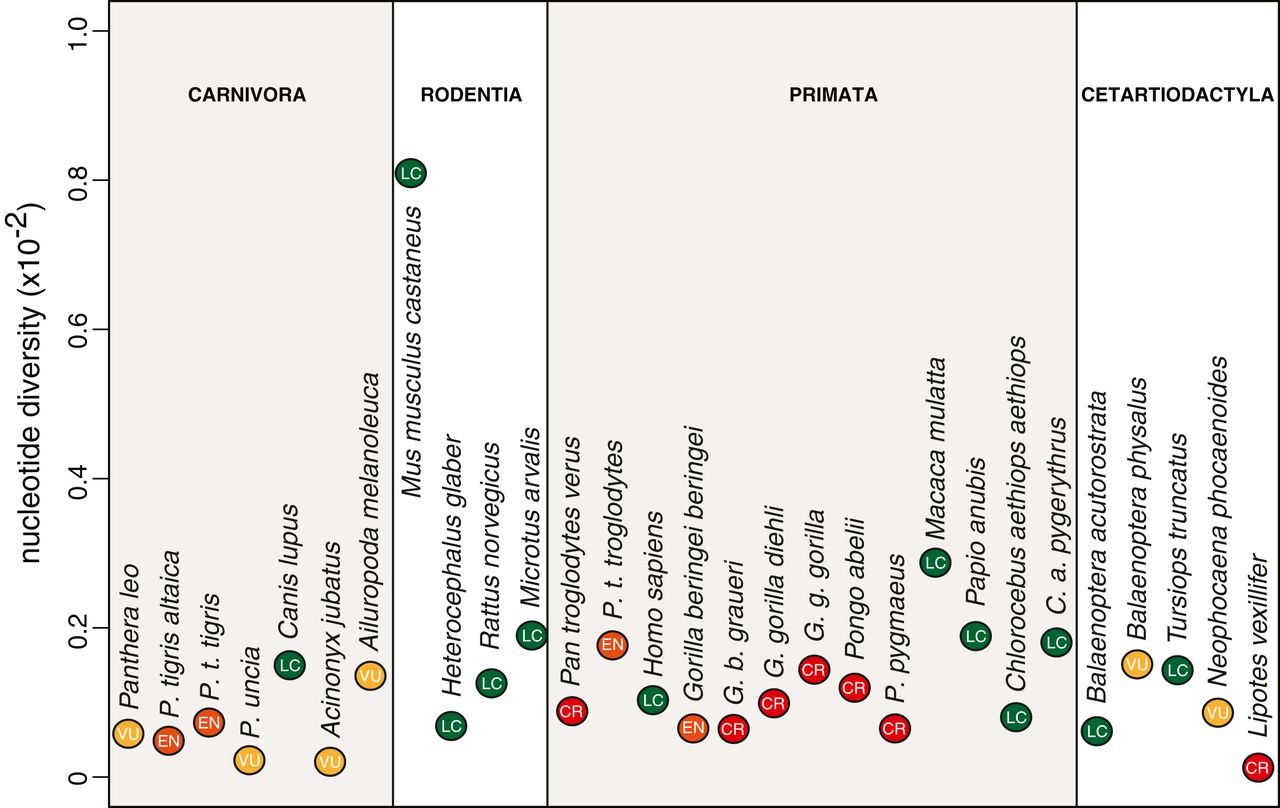
Their figure shows no relationship between nucleotide diversity and IUCN threat status, as other papers previously discussed. How the IUCN criteria has not been incorporating genetic diversity in their threat assessment is one thing, but to say that genetic diversity in the genomic areas that are neutrally evolving is less important for conservation than genetic diversity in areas that may directly affect fitness is another thing. The paper stated that both kinds of diversity will need to complement each other in explaining the evolutionary history and adaptability of species, but the call to emphasize functional aspects irks me to bring back the discussion around the peril of gene-targeted conservation. The discussion has been happening since people think only using MHC to do ex situ population management will be sufficient. Miller and Hedrick (1991) back then remind people about how some rare variants in MHC may actually be detrimental instead of beneficial; conserving only a few loci may not be a generalisable approach to all species.
Also, the lack of a relationship between genome-wide diversity and threat status does not dismiss the importance of genomic variation in conservation because the threat is not what makes low genomic diversity in the first place. A population’s genome-wide diversity is shaped by gradual or abrupt change in population size for a considerable number of generations of offspring. In other words, the population’s demographic history. This is why some threatened species have high genetic diversity; they are sometimes from captive populations with very few founders that are bred for quite a while, oftentimes with additional individuals from other captivity occasionally sent there, giving enough time for evolution to leave signs of recent population expansion. We may also not see any difference in different threat levels because species generation time is longer than the length of disturbance.
A study by Stoffel et al (2018), which is focusing on pinnipeds, explicitly tests this. In their analysis, they found a negative relationship between bottleneck probability and allelic diversity in various pinniped populations. Another study that was not so explicit in testing this but showing a similar trend was Feng et al (2019) who were looking at the relationship between effective population size and inbreeding coefficient. I take inbreeding coefficient as a proxy of recent demography.

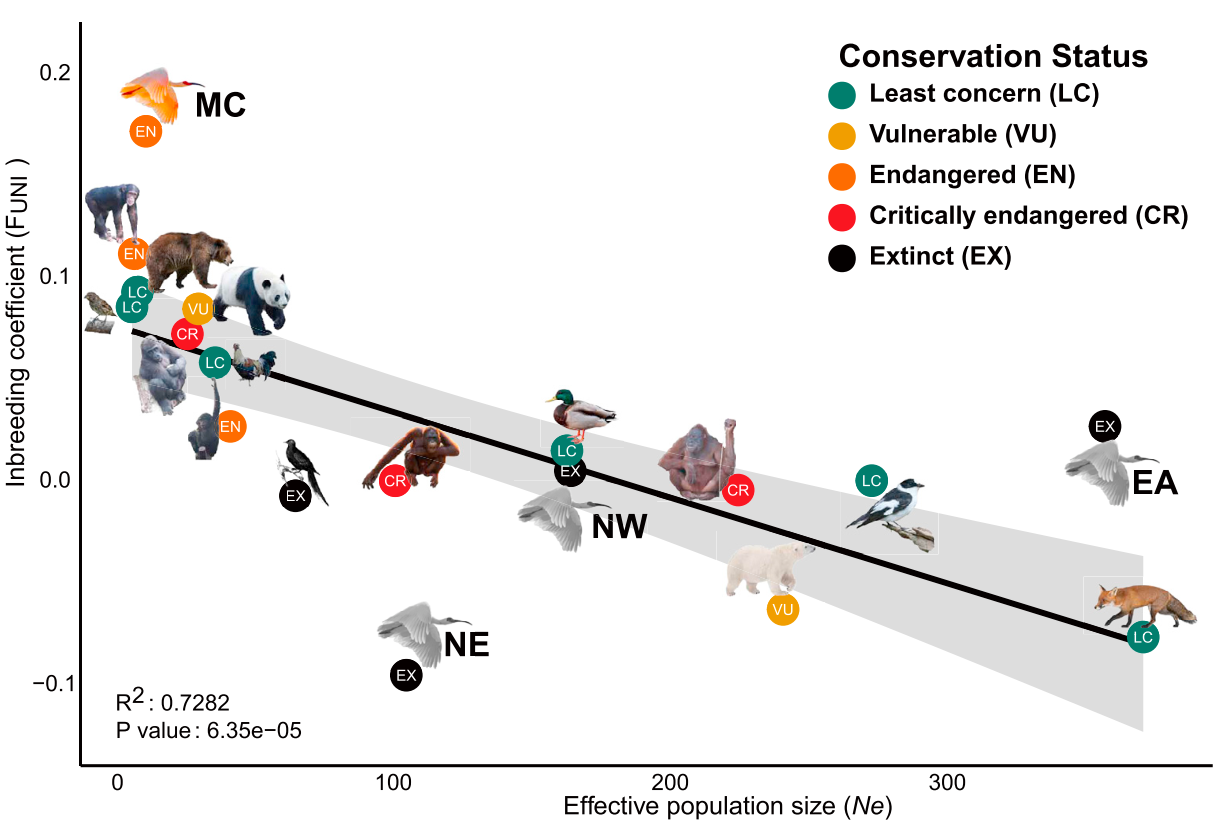
I believe the idea that genome-wide diversity is more affected by past demographic history is not new. It is also stated Diez-del-Molino (2018). I should also mention a thread by Marty Kardos’ Twitter account that has inspired this post:
Our current challenges with genome-wide diversity
Despite the ease of sequencing millions of base pairs of nucleotides within a day, the data we currently have is messy. Genome annotation of different species so far depends on ortholog sequences with some tolerable mismatches. In most cases we do not really know what a variant actually does in different species until we do a gene expression experiment or anything to prove that a mutation in a genome really does something. We could try to identify putative functional genetic variation by correlating the SNPs with environmental variables, but when no SNPs correlate with the environmental variables, can we say that these SNPs are not important for their long-term survival?
When we look at genome-wide diversity, I think it is important to assess a species’ past population history. When there is not enough information to look for variations that cause local adaptation, we can look for variations that may harm the populations in the long run, or what we often call the mutation load. In a commentary, van Oosterhout (2020) encourages conservation genomics to use information on the abundance of homozygous deleterious mutations and use the framework of “preventing loss of fitness” rather than “optimize fitness” framework.
Cataloguing deleterious variants may not always give a very straightforward story nonetheless. A book chapter by von Seth et al (2018) discovers no consistent patterns in the genome in extinct species such as mammoth, passenger pigeon, or cave bear. Genome-wide diversity can decline abruptly or gradually before extinction, or just being very low for a very long time before extinction. Of course the extirpation of species was caused by many more factors; genomics is just a piece of conservation science puzzle. A relevant paper about that, written by Lande (1988), explains this issue quite well.
I don’t think this means we should abandon calculating genome-wide diversity for conservation; we just need to be careful with what the results may or may not represent. The IUCN status contains a lot of variables that determine the extinction risk of the species, and although those variables are not directly related to genetics, they are as important as the genetic aspects themselves. The reason why we have to investigate genomics for conservation is exactly because there are still many questions around it.
All in all, everyone should choose their own “wars” to accelerate science, understanding faster what works and what does not work. Do you wanna go with functional or neutral variants? Let’s do them together and talk about what we found!