
This is the sixth in a series of posts where we explain the N50 (Nx) metric, discuss the problems surrounding it, give solutions to those problems, and suggest an alternative N50 metric for transcriptome assemblies.
Transcriptome assemblies are inherently different from genome assemblies. With genome assemblies most people strive to achieve an optimal contig length equaling an entire chromosome. Meaning that, if you have an organism with four chromosomes, your optimal (dream) genome assembly would consist of four long contigs. This is in stark contrast to transcriptome assemblies where contigs represent transcripts. An optimal transcriptome assembly will vary depending on your question, but would likely consist of all possible transcripts from all expressed genes, including alternatively spliced variants (isoforms).
Using the normal N50 metric for transcriptome assemblies can therefore be highly misleading, as transcriptomes do not strive to achieve long contig lengths and high N50, but instead one contig for each transcript. Furthermore, the most highly expressed transcripts do not necessarily constitute the longest ones and the majority of transcripts in a transcriptome assembly will normally have relatively low expression levels.

Fig 7. Example of a typical transcriptome assembly with a high number of short contigs and few contigs of intermediate length (from Senatore et al. 2015).
We strongly advise against using regular N50 metrics for transcriptome assemblies. Instead, other more appropriate measures can be used. The developers of the transcriptome assembler Trinity have invented the ExN50 metric, which takes into account the expression levels of each contig and is therefore a more suitable contig length metric for transcriptomes.
The ExN50 is calculated like N50 but is limited to the top most highly expressed transcripts that represent X% of the total normalized expression data (Fig. 8).
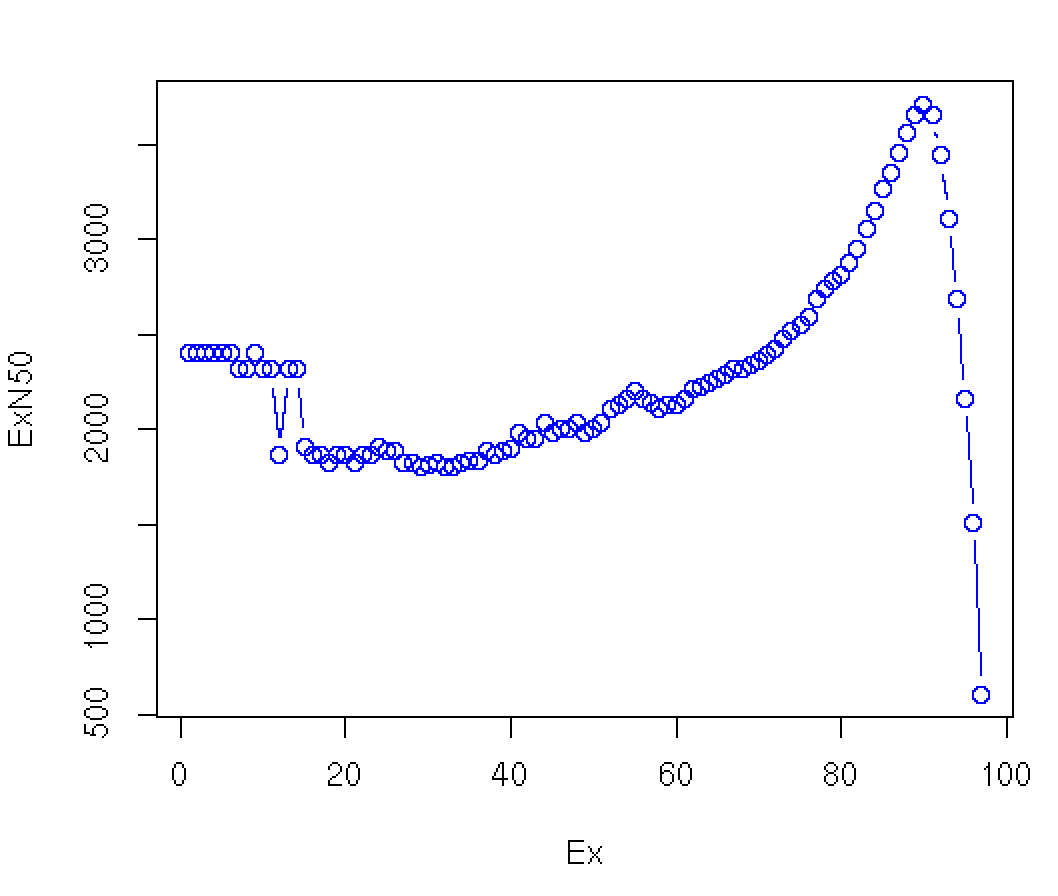
Fig 8. Plotting the Ex value against N50 shows that the E90N50 for this assembly yields the highest value and is a better indicator of assembly quality than the normal N50 metric. Figure derived from Trinity’s website.
There are other ways to evaluate the quality and completeness of transcriptome assemblies than using N50 measures. For example comparison of gene annotation to existing core gene databases. BUSCO is one such tool where you can compare your assembled transcripts to sets of core genes for specific groups of organisms (eukaryotes, bacteria, plants, etc).
rnaQUAST is a tool which aims to evaluate the quality of transcriptome assemblies by using both reference genome and gene databases, such as BUSCO. Transrate is another tool which evaluates transcriptome assemblies by inspecting contigs and mapping reads back to the assembly.
Just keep in mind when evaluating your assemblies to filter out contigs that don’t originate from the organism you are targeting, and to think critically about what N50 can actually tell you.
This was the final post in our little series about the N50 metric. If you enjoyed it, please let us know by leaving a comment or sharing the posts on social media. Thank you for reading!
This post has been jointly written by Elin Videvall, Andrey Prjibelski, and Alexey Gurevich.
Disclosure: Andrey and Alexey are developers of the rnaQUAST software.
References
Senatore A, Edirisinghe N, Katz PS (2015) Deep mRNA Sequencing of the Tritonia diomedea Brain Transcriptome Provides Access to Gene Homologues for Neuronal Excitability, Synaptic Transmission and Peptidergic Signalling. PLOS ONE 10(2): e0118321. doi:10.1371/journal.pone.0118321
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. doi: 10.1093/bioinformatics/btv351
Bushmanova E, Antipov D, Lapidus A, Suvorov V, Prjibelski AD (2016) rnaQUAST: a quality assessment tool for de novo transcriptome assemblies. Bioinformatics, btw218.
Smith-Unna R, Boursnell C, Patro R, Hibberd JM, Kelly S (2016) TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Research. doi: 10.1101/gr.196469.115