
This week I’ve invited a good friend and fellow Homo sapiens, Jacob Tennessen, to contribute a guest post to the Molecular Ecologist. Jacob is a Postdoctoral Scholar at Oregon State University, where he currently works with Mike Blouin and Aaron Liston, but he has also worked at the University of Washington with Josh Akey. It was while he was up in Seattle that Jacob made some profound discoveries that are relevant both to us as a species and as Molecular Ecologists. I’ve invited Jacob to share some of his insights into this fascinating topic…

The term “model species” or “model organism” usually refers to critters like Drosophila that are conducive to basic experimental biology and can yield results relevant to other taxa, including humans. At least, that’s how it gets justified in NIH grant applications: you study non-humans to learn about humans, not the other way around. In molecular ecology, though, the tables have turned. Molecular ecologists study non-humans using methods that were initially tried on humans, and interpret their data in a framework of theoretical population genetics that has been validated with human genetic data. Thus, humans are actually a model species for molecular ecology. Unfortunately, not all molecular ecologists fully appreciate the importance of human genetics to their discipline.
Molecular ecology is essentially population genetics of wild, ecologically interesting species. Because these species are generally difficult to breed or otherwise manipulate in the lab, molecular ecology is often observational rather than experimental. Non-invasive genetics and “natural experiments” are employed to make inferences about evolutionary history, behavior, fitness, and other aspects of natural history. These same restrictions also apply to humans: breeding humans in the lab is as ethically fraught as it is logistically challenging. But, the difference between studying humans and, say, elephant seals is that the established knowledge base for humans is much greater. The combined size of available population genetic datasets in humans is a billion-fold larger than for most species, even some that have already been the target of molecular ecology studies, and these human data are much better annotated and validated. Furthermore, being humans ourselves, we all have an intuitive grasp of the human phenotype, so statements about, say, human genetic diversity are readily placed in the familiar framework of human phenotypic diversity.
Thus, the field of human population genetics has always been a step or two ahead of the molecular ecology of wildlife. Common techniques like mitochondrial- or microsatellite-based phylogeography analyses were pioneered with data from humans. Research into human molecular ecology has yielded countless fascinating stories that provide a baseline for what to expect when examining other taxa. Some are well-known textbook examples, like the sickle-cell hemoglobin balanced polymorphism that conveys resistance to malaria, or the human global diaspora reflected in sequence diversity that traces back to “mitochondrial Eve” and “Y-chromosome Adam.” There are also many clear examples of how individual genes have contributed to geographically local adaptation: LCT alleles allowing adults to digest milk in regions of Europe and Africa where dairy agriculture was historically important, SLC24A5 reducing skin pigmentation in Europe, EPAS1 allowing Tibetans to breath easily at high altitudes, and plenty of others. Human DNA can even reveal historical patterns of behavior. For example, African hunter-gatherer cultures tend to be matrilineal while herding cultures tend to be patrilineal, as inferred from contrasting dispersal patterns in Y chromosome and mitochondrial DNA.
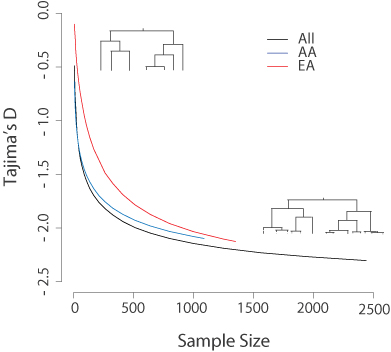
To learn what the hot new methods in molecular ecology will be, look to the human genetics literature. For example. the human Exome Sequencing Project demonstrated the use of targeted capture for population genetics, and this handy way of generating partial genomic datasets is ideal for other studies, including recent investigations of speciation in whitefish and butterflies. Our ESP data confirmed that for a rapidly expanding species like humans, measurements of the site frequency spectrum such as Tajima’s D will severely underestimate the rate of growth unless the sample size is quite large (N>1000). Molecular ecologists should take note, because Tajima’s D is a popular statistic and few real populations are at perfect mutation-drift equilibrium (see figure below).
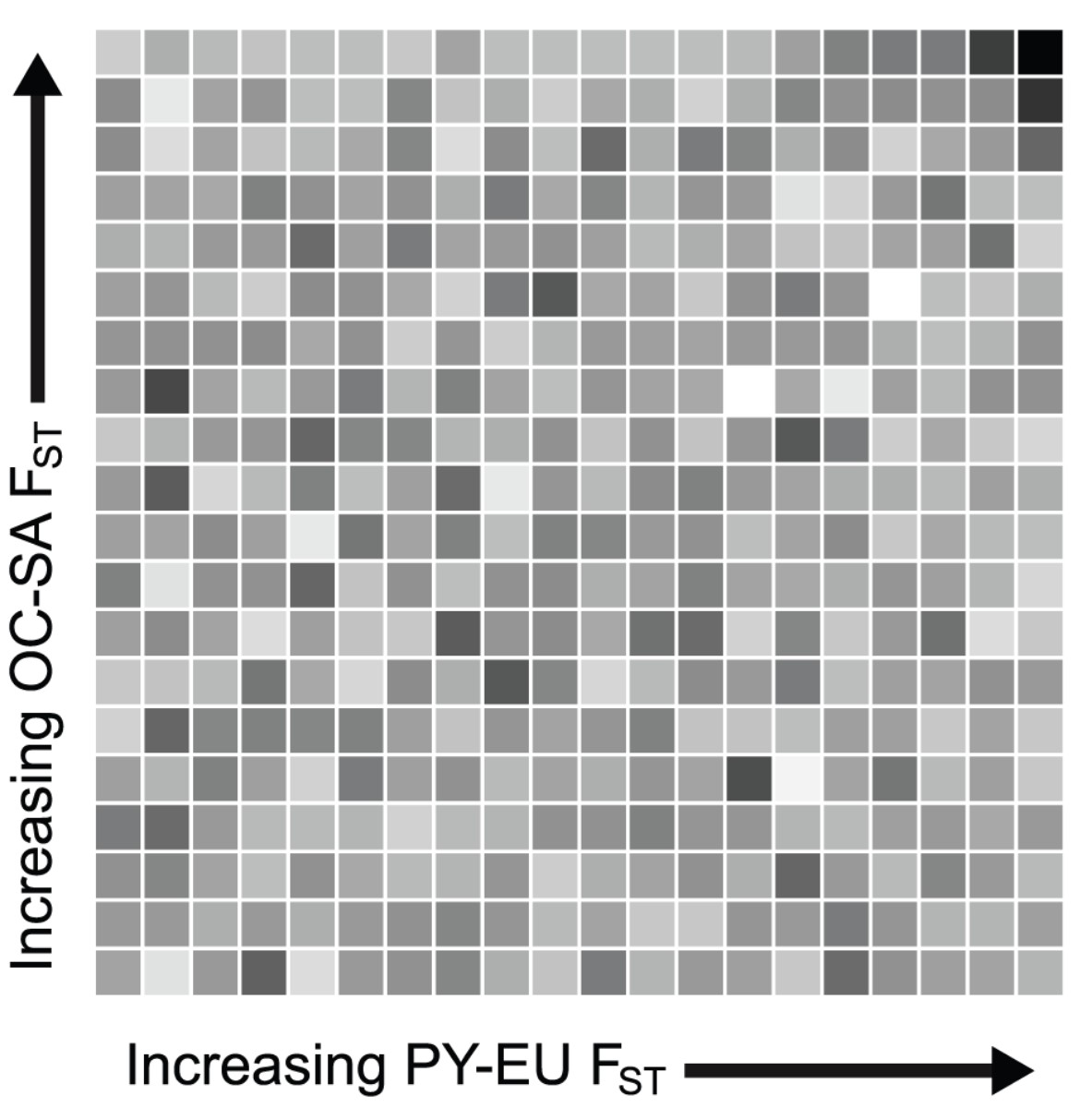
Human genetic data is also a fertile testing ground for new statistical analyses. With Josh Akey, I showed that the same genetic variants evolve in parallel in independent human populations in response to local selection pressures. The test we devised is conceptually simple, and it would be great to see it applied to other species with heterogeneous ranges. In short, we start with the popular “outlier FST” approach in which genetic markers showing unusually high divergence between populations are assumed to be responding to divergent selection. It can be challenging to conclusively show that this divergence is really driven by selection and not just the normal tail of a neutral distribution. However, if an unexpectedly high proportion of markers show high divergence in population pair A and B as well as in phylogenetically independent population pair C and D, neutrality can be firmly rejected (see figure below).
Test of parallel divergence. For >26,000 SNPs, we calculated FST between African Pygmies and Europeans (PY-EU) and between Oceanians and South Americans (OC-SA). SNPs were ranked by each FST measure and partitioned equally into 5% bins. For each square, darker shades represent a higher proportion of SNPs falling in that particular sector (~50-100 SNPs per sector). The dark sector in the upper right corner contains more SNPs than any other sector; that is, there is an unusually high number of SNPs showing extremely high FST in both independent comparisons. Divergent natural selection acting independently both between Europe and Africa and between Oceania and South America is the best explanation for this observation. From Tennessen and Akey 2011.
The list goes on. Numerous useful bioinformatic tools have been developed for human genetics but remain underutilized in other systems. In particular, I recommend the Perlymorphism suite of scripts in BioPerl, ∂a∂i for inferring demographic history, Bayenv for finding genes underlying local adaptation to specified environmental variables, and VCFtools and related software for vcf format genotype files.
So, what are the most important things we have learned from studying our own molecular ecology? Perhaps the primary lesson from human population genetics is that intergroup differences that seemed substantial to our subjective brains, like between Africans and Europeans, turned out to be minor. There are few if any fixed autosomal differences between continental groups, and the phenotypic markers we are inclined to use, like skin color, are encoded by some of the most divergent loci, making them a poor proxy for overall evolutionary distance. A related major lesson is the surprising ubiquity of “soft sweeps,” or positive selection acting on standing variation. Unlike the classic model of a newly arisen mutation rising quickly to fixation, most geographically local adaptation in humans comes from more subtle changes in the frequency of existing alleles, hence the dearth of fixed differences. A third lesson is that the most genetically diverse human populations are found in our ancestral homeland in sub-Saharan Africa, with basal populations such as the San showing particularly high polymorphism.
The implications of these lessons are illustrated in the following thought experiment. Imagine that some horrific natural disaster killed all humans except a small number of San individuals. Numerous human phenotypes would be lost, but the alleles underlying many of these phenotypes would be present at low frequencies, and the prevalence of parallel evolution via soft sweeps suggests that common traits would readily re-evolve given conditions in which they are adaptive. Thus, although the human cost would be immeasurable, the genetic cost would be slim. This scenario is somewhat counterintuitive, and it’s worth bearing in mind when making management decisions about other species with respect to conserving genetic diversity.
The methods and results of human population genetics continue to trickle down to the scientists studying wild flora and fauna. But there is no reason it has to be a trickle. Turn on the proverbial tap and find out what your own molecules can tell you about the ecology of your favorite organism.